

/image%2F1914771%2F20160306%2Fob_ccee94_tpe-tyrosinemie-de-type-1-au-saguenay.PNG)
La tyrosinémie de type 1 est une maladie génétique qui entraîne un défaut de FAH et une accumulation de métabolites potentiellement toxiques... De plus, elle atteint de nombreux individus, aboutissant ainsi à un réel problème de santé public.
A- Les origines de la tyrosinémie du génotype au phénotype moléculaire
En 1999, 34 mutations sur le gène FAH, localisé sur le chromosome 15, ont été identifiées :
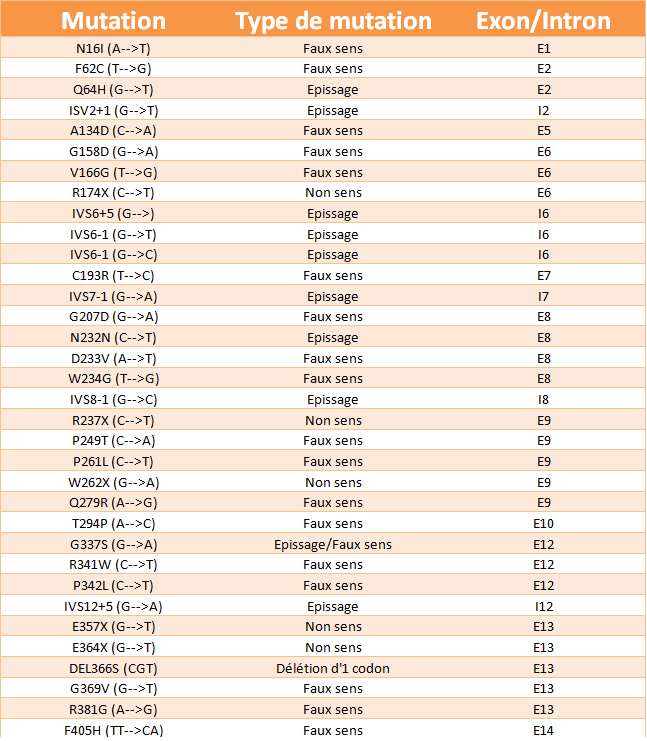
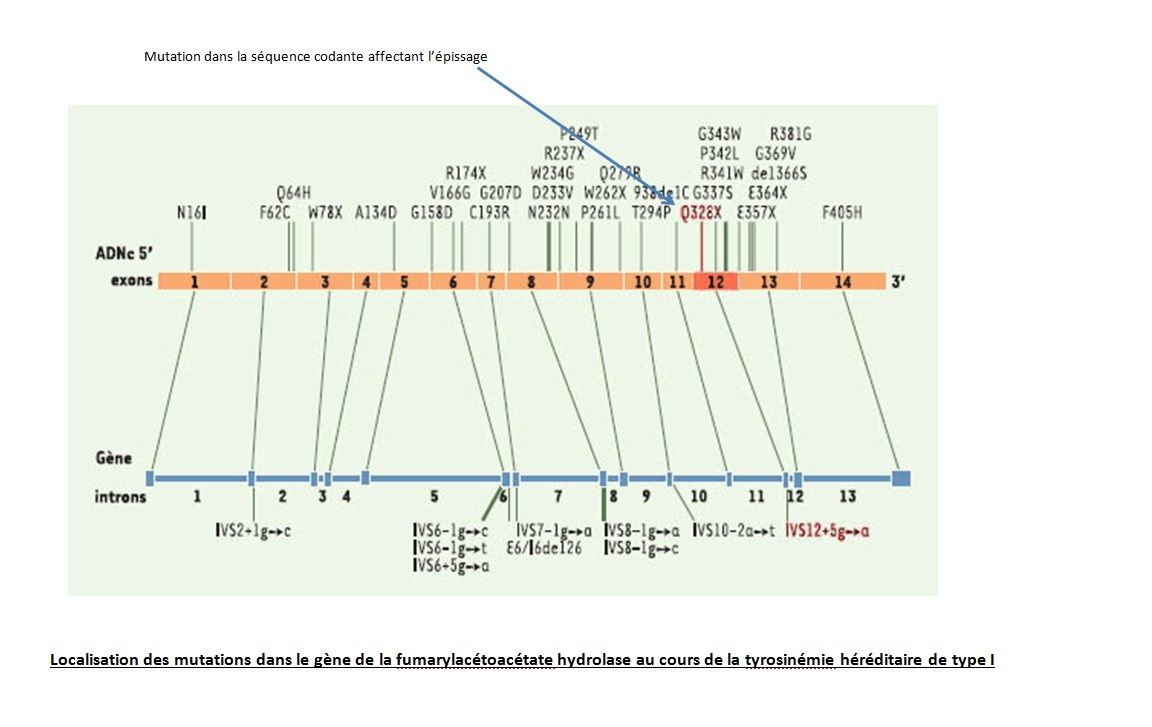
Tableau réalisé par le groupe
Ces mutations sont dispersées aléatoirement sur le gène et donnent différents types de mutation et donc des génotypes différents :
- la mutation faux-sens : mutation due à des altérations dans la séquence ADN changeant un acide aminé en un autre. Cette mutation est nommée par la lettre d'abréviation de l'acide aminé normal - puis le numéro du codon - et enfin la lettre abréviation de l'acide aminé muté .
- la mutation non-sens : mutation due a des altérations dans la séquence ADN qui contient un codon stop , de sorte que la protéine est tronquée. Cette mutation est nommée par la lettre abréviation de l'acide aminé normal – puis le numéro de codon – et enfin X , signifiant le codon stop.
- mutation épissage : mutation qui se produit dans les régions entre les exons (les régions codantes). Les introns sont cependant également importants pour l'épissage de l'ARNm. Un changement d'une seule base, une insertion ou une deletion peuvent provoquer un mauvais épissage de l'ARNm et conduire à une protéine anormale . Cette mutation est nommée IVS (intrvening sequences) – numéro de l'introns - emplacement de la mutation sur l'intron par rapport à l' exon le plus proche ( + si en amont , - si en aval ).
Cependant dans 95% des cas, la mutation IVS12+5 (G-->A) en position 1009 qui est une mutation non-sens, est la cause de la tyrosinémie de type I. Suite à cette mutation, le gène subit un épissage alternatif bien particulier qui est obtenu à cause d'une substitution du nucléotide guanosine en adénosine en positon 5 sur l'intron 12. Cet épissage est composé de trois bandes :

Schéma réalisé par le groupe
- une bande S tronquée lors de l'épissage qui présente une délétion de 102 nucléotides de la position 1017 à 1118. De ce fait 34 acides aminés, de la position 321 à 354, ont été supprimés.

Schéma réalisé par le groupe
- une bande M ne présente seulement que quelques changements qui ne modifient ni sa taille, ni son fonctionnement.

Schéma réalisé par le groupe
- une bande L plus longue que la normale présentant une insertion de 105 nucléotides entre la position 1118-1119. De plus cette insertion contient un codon stop, ainsi la protéine finale est tronquée après 320 acides aminés.

Schéma réalisé par le groupe
L'origine de la tyrosinémie de type 1 relève donc du hasard.
Nous nous concentrons donc sur les conséquences de ces changements du génotype sur le phénotype moléculaire :
Cette mutation donne lieu à une déficience du gène aboutissant à la production de l'enzyme fumarylacétoacétate hydrolase (FAH) une enzyme de la chaîne de catabolisme de la tyrosine indispensable à sa destruction. Nous pouvons observer les différentes étapes de cette chaîne destructrice de la tyrosinémie ci-dessous :



Molécule construite par le groupe

Molécule construite par le groupe

Molécule construite par le groupe
B- Du phénotype cellulaire au phénotype macroscopique
Ces modifications du phénotype moléculaire ont des conséquences sur le phénotype cellulaire:
les individus atteints de la tyrosinémie de type 1 manquent de FAH (en majorité localisé dans les reins et le foie) ce qui entraîne une HYPERTYROSINEMIE, ainsi qu'une accumulation de certains métabolites dans ces organes: la fumarylacétoacétate (FAA) et le maléylacétoacétate (MAA) qui, après décarboxylation engendrent la succinylacétone : une toxine. Cette enzyme est normalement absente chez l'homme, sa détection permet donc de diagnostiquer la tyrosinémie de type 1.
- Le FAA peut, en trop importante quantité, engendrer des réactions pouvant aggraver la tyrosinémie. En effet, il est un agent génotoxique : il est dangereux pour la santé et possède la capacité d’altérer de manière directe le matériel génétique :
- en entraînant des anomalies du fuseau mitotique, des défauts de ségrégation des chromosomes en anaphase et donc la formation de cellules multinuclées et de micronoyaux. Ainsi, le FAA provoque des anomalies dans le nombre de chromosomes (aneuploïdie), et donc il a un effet aneugène.
- en provoquant des mutations au niveau du matériel génétique (37% de cancer du foie chez les patients atteints). Cet effet mutagène est potentialisé par un arrêt du cycle cellulaire en G2/M dû au FAA, suivi d’une entrée en apoptose => ces perturbations du cycle cellulaire sont dépendantes de la concentration de glutathion (GSH) intracellulaire sauf que...
- ... le FAA réduit la concentration cellulaire de glutathion qui joue un rôle important dans la détoxification des espèces réactives de l'oxygène (comme la tyrosine) => protégeant ainsi les cellules contre leurs effets toxiques et/ou mutagènes.
Enfin, toutes ces modifications néfastes à la santé de l'homme aboutissent à deux types de phénotypes macroscopiques malades dont nous avons déjà fait la description dans le C- de la partie 1
C- Un problème de santé publique indéniable et requérant des traitements particuliers
Un problème de santé publique est la détérioration de la santé d’une partie de la population, qui, par : sa gravité, son nombre de victimes et son coût, nécessite une intervention de l’Etat.
Ainsi, depuis plus de 30 ans, un ensemble de ressources et d’activités est déployée pour dépister et prendre en charge, le plus tôt possible, tous les nouveau-nés québécois atteints de phénylcétonurie, d’hypothyroïdie congénitale ou de tyrosinémie de type I.
Pour ce qui est de la tyrosinémie, la survie des enfants atteints est réduite et dépend de l’âge d’apparition des symptômes. Afin de remédier à cela, le ministère de la Santé et des Services Sociaux (MSSS) a mis en place :
- une prévention primaire qui agit en amont de la maladie, elle a pour but de diminuer l’apparition des nouveaux cas de maladie. Elle agit sur les facteurs de risques de la tyrosinémie en permettant aux parents désirant un enfant et habitant au SLSJ de passer un test de risque génétique (calcul de probabilité d’avoir un enfant malade) en fonction de leurs antécédents familiaux de cas tyrosinémiques
- une prévention secondaire qui a pour but de dépister la maladie pour mieux pouvoir la soigner. Le dépistage de la tyrosinémie repose principalement sur le dosage biochimique de marqueurs contenus dans un échantillon sanguin prélevé à la naissance et séché sur papier buvard. Cependant, l’accessibilité à ce test est moins évidente pour les nouveau-nés hors des hôpitaux et cliniques québécois (ex : à domicile) ; les enfants de l’adoption internationale ; les enfants de parents immigrants
- une prévention tertiaire qui vise à éviter les rechutes ou la chronicité de la maladie. Elle se concrétise par des traitements efficaces précoces. Pour le cas de la tyrosinémie, les traitements sont : une diète (un régime alimentaire faible en protéines animales et végétales durant toute la vie => les personnes atteintes de tyrosinémie peuvent obtenir gratuitement des produits à faible teneur en tyrosine dans le cadre d’un programme entièrement financé par le ministère de la Santé et des Services sociaux), ET un médicament, le NTBC (expérimenté depuis 1994, ce médicament empêche l’accumulation des déchets les plus toxiques et les plus dommageables pour le foie, les reins et les nerfs).
Dans certains cas, il est possible d’opérer une transplantation hépatique afin de remédier aux problèmes du foie et aux crises neurologiques mais cette intervention chirurgicale est risquée et nécessite un traitement immunosuppresseur à vie pour éviter le rejet du nouveau foie.
Cependant, ces traitements permettent d’améliorer le quotidien des malades, pourtant ne sont que des traitements symptomatiques, ils ne guérissent donc pas la maladie.
C'est pourquoi l'information de la population, le calcul de risque génétique, et les test prénataux sont si importants afin de réduire la prévalence de la maladie.
Le coût de dépistage et la prise en charge des malades est très élevé pour l'Etat mais à long terme, les bénéfices sont évidents : les dépenses occasionnées seront compensés par la diminution du nombre de malade de génération en génération. En quelques chiffres: le coût d'un traitement annuel de NTBC (pour un patient de 45 kg) varie de 7 500 $ à 70000 $CA en fonction de sa durée de vie et si la maladie est plus ou moins développée...
BILAN
Ainsi, le Québec doit faire face à un problème de santé majeur et onéreux à cause d'une maladie autosomale récessive et mortelle sans traitement précoce: la tyrosinémie de type 1. La tyrosinémie de type 1 est due à une mutation du gène codant la FAH. Cette enzyme, dans le cas d'un individu sain, détruit la tyrosinémie. Mais dans le cas d'un malade, la FAH n'est pas produit, donc il ne catabolise pas la tyrosine (en majorité localisé dans les reins et le foie) ce qui entraîne une HYPERTYROSINEMIE, plus, une accumulation de métabolites dans ces organes: le maléylacétoacétate (MAA) et le fumarylacétoacétate (FAA) qui est très toxique pour l'organisme. Par conséquent, les individus malades doivent être pris en charge très tôt. Mais les mesures de prévention et de suivi des malades mises en place, permettront en principe de faire baisser la prévalence de cette maladie au fil des générations puisque les gènes défectueux seront de moins en moins transmis.
Nous pouvons alors nous demander quelles sont les causes des aberrations génétiques propres au Saguenay-Lac-Saint-Jean et donc pourquoi la tyrosinémie de type 1 est plus fréquente dans cette région que dans le reste du monde.
Ci-dessous, une vidéo pour les courageux très intéressante et synthétique sur le phénotype moléculaire malade et les traitements qu'ils nécessitent... Mais en anglais! Immergez-vous en Amérique du Nord!